Retinoblastoma
General Information
Retinoblastoma is of neuroepithelial origin, arising from the nucleated photoreceptor layers of the inner retina of one or both eyes. The disease consists of small round blue cells with scant cytoplasm and large nuclei. Calcification commonly occurs in necrotic areas. Pathologically Flexner-Weinersteiner Rosettes (neuroepithelial rosettes) are described microscopically. The Flexner-Weinersteiner Rosettes are an attempt to differentiate into photoreceptor cells.
In addtion to the Flexner-Weinersteiner Rosettes, the cells appear as small round blue cells, with calcifications and necrosis.
As retinoblastoma grows, it may cause retinal detachment or invade the vitreous humor. The tumor may also form a pedunculated mass as an exophytic growth. It is not unusual for the vitreous to be seeded and tumors of the vitreous grow despite the lack of blood supply.
| Flexner Weinersteiner Rosette of Retinoblastoma |
|---|
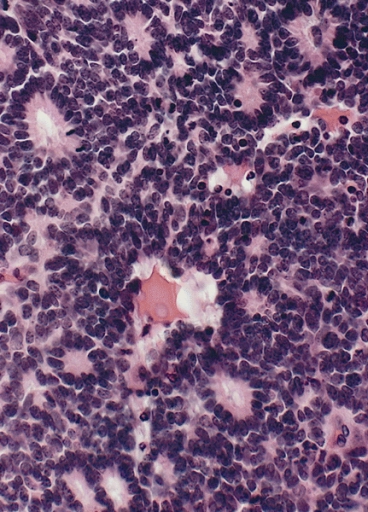 |
Epidemiology
Retinoblastoma is the most common intra-ocular malignancy of childhood. There are about 250 - 300 case per year of retinoblastoma. 95% are less than 5 years old. Familial retinoblastoma cases are less than 1 year old, and 2 years old for sporadic cases. Metastatic leukemia to the eye is the most common tumor, but retinoblastoma is the most common primary disease of the eye.
For all age groups, the three most common ocular tumors are:
- Metastatic disease to the eye
- Melanoma
- Retinoblastoma
Retinoblastoma shows no predilection for sex, race or sidedness. Between 60 - 85% of all cases are unilateral. The mean age of diagnosis is 2 - 4 months and diagnosis is usually made before 3 years of age. Unilateral retinoblasatoma is diagnosed at a later age than bilateral retinoblastoma. A Mayo Clinic report noted that median ages at diagnosis were 4.5 months for bilateral disease and 22 months for unilateral disease.
About 20% - 30% are bilateral or multifocal. 70% - 80% are unilateral (depending on the data set -- academic centers may report a higher rate of bilateral disease as tertiary referral centers). 40% of multifocal retinoblastomas are hereditary but only 10% have a positive family history. 60% are from new germline mutations: 93% have a negative family history.
Patients with retinoblastoma are also prone to osteosarcomas.
- Between 60% and 85% of all cases are unilateral
- About 20% - 30% are bilateral or multifocal
- Bilateral tumors are typically multifocal
- Only 10% have a positive family history but 40% are inheritable. 60% are new germline mutations
- 93% have a negative family history
- Mean age at diagnosis is 2 -4 months and usually before 3 years of age.
- Unilateral disease is diagnosed at a later age than bilateral retinoblastoma
- Unilateral median age of diagnosis (Mayo) 22 months
- Bilateral median age of diagnosis (Mayo) 4.5 months
Genetics
The RB1 tumor suppressor gene is mutated in retinoblastoma. There is a 2 hit theory of initiation of disease:
- First hit: germline deletion
- Second hit: somatic mitotic recombination error
The RB suppressor gene is found on chromosome 13. Various mutations are seen in each of 20 exons but not mutational hot spots are demonstrated on the RB gene.
Retinoblastoma Genetics: the 2 Hit Hypothesis
This hypothesis is based on the fact that the Rb gene is autosomal recessive. The RB gene is identified on 13q14.
In the case of unilateral disease, it is generally non-hereditary and results from somatic mutations of both Rb genes in the same cell. Therefore, only one eye will have the disease.
In the case of bilateral disease, the egg or sperm carries a mutant gene. Then the cell attains a second mutation. This cell then divides into retinal layers for both eyes.
| Toma et al, Br. J. Opthalmol. 1995;79:112-117 |
Patterns of Spread/Natural History
There are five identified patterns of spread.
- Local extension of disease into vitreous
- Tracking the optic nerve to the brain
- Spread via CSF to the leptomeninges or subarachnoid spaces
- Hematologic metastases
- Lymphatics from the conjunctiva, ciliary body and iris.
The most common sites of hematogenous spread in retinoblastoma are to the bone, liver and spleen. These metastases are seen in 10% - 15 % of patients at presentation. Thickness of the tumor relates to the propensity to track the optic nerve to the brain, invasion of the uvea , orbit and choroid. The size of the tumor also correlates to an increased risk of metastases.
Trilateral Retinoblastoma is bilateral retinoblastoma and a CNS mid-line PNETinvolving the pineal gland. representing 3% to 9% of inheritable retinoblastoma. It arises from residual ectopic retinal tissue in the pineal gland and is uniformly fatal.
Prognostic Factors
The main prognostic factors are:
- Delay in diagnosis beyond 6 months of age
- History of intra-ocular surgery (do not biopsy!) leading to seeding, cataracts and thick tumors
- History of radiation therapy because such patients are usually locally advanced.
Workup and Staging
Presenting Symptoms
Patients in the developed nations usually present with (in order):
- leukocoria followed by
- strabismus followed by
- painful glaucoma
- irritability.
In developing countries, the presentation is usually more advanced with proptosis, orbital mass and metastases.
Patients presenting with leukocoria may have congenital cataracts, hyperlastic primary vitreous, retrolental fibrodysplasia, Coat disease, toxocariasis and toxoplasmosis. There are all on the differential diagnosis.
Biopsy is generally not done for retinoblastoma to avoid the risk of seeding. The diagnosis is established clinically, based on clinical and imaging findings.
Retinoblastoma typically presents with a white pupillary reflex (leukocoria), which may be discovered in a photograph with an abnormal light reflex or with an opthalmoscope at a physical examination. Large retinoblastomas produce the white reflex when the flash bounces off of it or when the tumor produces retinal detachment.
| J Morley-Smith (public domain), 2008 |
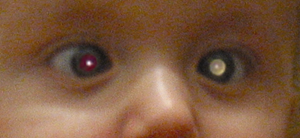 |
The workup for an intraocular mass includes the History and Physical, an examination under anesthesia with maximum dilated pupil, labs, and imaging consisting of ultrasound, MRI and CT. The retinal examination may reveal a raised white or white-pink-yellow mass with tortuous vessels. 90% of retinoblastomas are calcified.
| J Morley-Smith (public domain), 2008 |
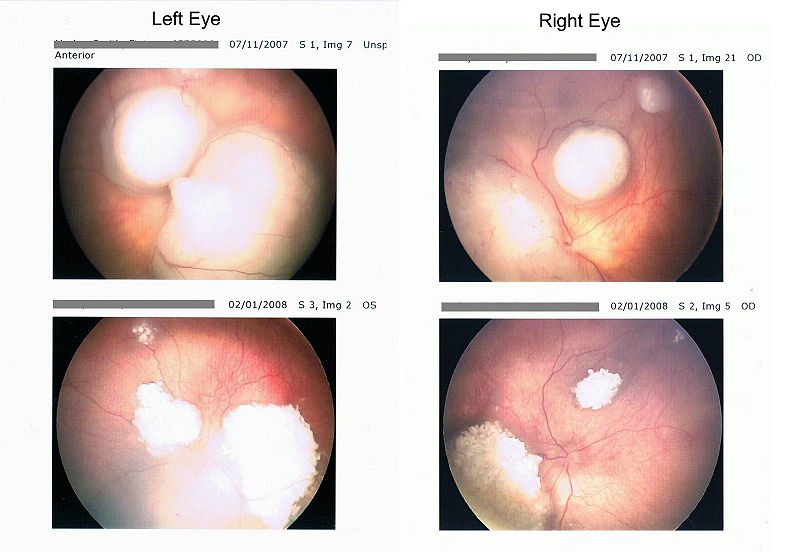 |
Bone scans are indicated when the mass is found on localized imaging to extend beyond the globe with deep invasion. If this is the case then a bone scan, bone marrow biopsy and lumbar puncture are necessary to assess extent of disease.
Staging
Reese Ellsworth Classification
The Reese Ellsworth Classification system is a good system when used for external beam radiotherapy treatment decisions. It is a 5 group system which is established for prediction of ability to maintain sight after external beam radiaton therapy. This classification system does not predict survival. The Reese-Ellsworth Staging system:
| Group I: | Very favorable for the maintenance of sight
|
| Group II: | Favorable for the maintenance of sight
|
| Group III: | Possible for the maintenance of sight
|
| Group IV: | unfavorable for the maintenance of sight
|
| Group V: | very unfavorable for the maintenance of sight
|
International Classification for Intraocular Retinoblastoma
This staging system is presently used by all ongoing COG protocols. The International Classification Scheme for Intraocular Retinoblastoma is used to classify outcomes related to chemotherapy administration and was developed by COG and the Children's Hospital of Los Angeles. It too is a five group staging system.
| Group A | Small intra-retinal tumors away from the fovea and disc
|
| Group B | All remaining discrete tumors confined to the retina
|
| Group C | Discrete local disease within minimal subretinal or vitreous seeding
|
| Group D | Diffuse disease with significant vitreous or sub-retinal seeding
|
| Group E | Presence of any of the following poor prognosis features:
|
Treatment
The primary goal of therapy is cure, with vision preservation being a key secondary goal. Retinoblastoma infrequently metastasizes, therefore the chance of cure is high. Enucleation is recommended only in a blind eye or for which there is no reasonable expectation of sight after local therapy. The general treatment paradigm for retinoblastoma is aimed at vision preservation in the case of unilateral intra-ocular retinoblastoma. The goal is to preserve the eye with chemotherapy reduction x 6 cycles of vincristine/carboplatin/etoposide (VCE). This is followed on by focal therapy if necessary. Focal therapies include enucleation, External Beam or Brachytherapy (plaque) radiation therapy, cryotherapy, laser photocoagulation, thermo-chemical ablation, sub-Tenon injection of carboplatin. The capsule of Tenon is the thin outer membrane enveloping the eye.
Focal Therapies
Enucleation
Enucleation is indicated in unilateral retinoblastoma where the eye is blind. In bilateral retinoblastoma where both eyes are blind, a bilateral enucleation is performed. Enucleation may be performed as salvage when the tumor can no longer be controlled with alterantive, more conservative measures.
Risk Factors for Metastases at time of Enucleation
Risk factors for metastases can be identified at the time of encleation. These include
- Optic Nerve invasion
- The presence of tumor beyond the lamina cribrosa is a risk factor for metastatic disease due to access to the subarachnoid space which allows CSF dissemination to the spinal cord and brain. Optic nerve invasion is present in about 1/3 of enucleated globes and often correlates with choroidal or scleral invasion.
- Optic nerve invasion is characterized by preliminary, laminar, post-laminar and up to the line of transection. When the optic nerve was not involved, disease free survival was 97%. When the optic nerve was involved up to the line of transection, the DFS was 55%.
- Uveal invasion
- Choroidal invasion allows access to the scleral and emmisary veins but is difficult to quantify. When optic nerve invasion is excluded, there is no clear risk of distant metastases, but a trend was still present with choroidal invasion with p=0.10.
- Orbital Invasion
- When the orbit is invaded access to the lymphatics and vascular channels is gained. This creates a significant risk for metastatic disease and is considered an indication for aggressive local and systemic treatment. Microscopic orbital extension cannot be detected by clinical examination. Pre-operative studies, such as MRI or ultrasound may show larger extensions.
- Choroidal Involvement
- Choroidal involvement is considered a poor prognostic sign. Hematogenous spread of retinoblastoma occurs via the choroidal vessels, but choroidal invasion is not rare and distant metastases are rare. It appears that the correlation between distant metastases and choroidal invasion is associated with the volume of choroidal invasion.
Exenteration
Exenteration is used where there is frank tumor beyond the globe. This is usually followed by post-operative radiation therapy and chemotherapy.
Other local therapies
Cryotherapy is useful for small lesions at least for disk diameters from the fovea and optic disc.
Treatment By Stage/Selection and Techniques of Therapy
The generally accepted techniques of therapy by international retinoblastoma groupings:
- Group A: small intra-retinal tumors away from fovia/disc: focal therapy only (laser, cryo, hyperthermia, brachytherapy)
- Group B: tumors larger than 3 mm or close ot fovia/disc: VC chemotherapy x 6 cycles → focal therapy
- Group C: discrete local disease with minimal vitreous/subretinal seeding: VCEx6 → focal therapy
- Group D: Diffuse disease confined to globe: VCEx6 → external beam RT
- Group E: Enucleation → VCE
Radiation therapy is used in those cases where there is disease persistence or progression on chemotherapy, in those cases with extra-ocular extension or tumor within the macula. Radiotherapy is also used in diffuse vitreous seeding or subretinal implants. Radiation therapy is only used if vision preservation is possible.
The evidence supporting the use of radiation therapy is a retrospective review of chemotherapy ± local treatment in eyes with seeding prior to treatment. Shields reported that the addition of local treatment to chemotherapy reduced the incidence of sub-retinal and vitreous recurrences from 75% to ZERO percent. for vitreous seeding and 67% to 0% for sub-retinal seeding.
Adjuvant chemo-radiation therapy is indicated in enucleation with the presence of positive margins or positive lymph nodes, post enucleation.
Radiation Therapy Techniques and Doses
External beam radiation therapy doses commonly used are 36 - 40 Gy with progressive disease and 26 Gy with persistent disease after VC or VCE chemotherapy. the lower dose is used after sub-Tenon chemotherapy because of the increased risk of retinopathy with chemotherapy.
In the presence of orbital disease, (ie extra-ocular disease) radiation therapy to the orbit, chemotherapy ± radiation therapy for palliation, intrathecal chemotherapy and high dose chemotherapy with stem cell transplant are used. Eye preservation rates in Reese-Ellsworth I-III are 65% - 90% and in IRBG, 60% - 90%.
External beam radiation therapy is generally IMRT or 3D conformal treatments with 4 - 6 MV photons to the entire globe + 5 - 8 mm of optic nerve extension. For lower Reese-Ellsworth stages, the iris and lens may be spared. 0.5 cm bolus is used if required for dosimetry. Fields are usually 4 field obliques, proton beams or IMRT. More recently a report by Reissner indicated IMRT provided superior coverage when intercompared with other photon/electron techniques. For bilateral disease, opposed lateral beams supplemented by anterior obliques are used, or in the alternative, IMRT radiation therapy.
Bilateral retinoblastoma treatment is individualized for each eye.
Brachytherapy using an I-125 or Ru-106 eye plaque may be used, provided the tumor is a solitary lesion at least 3 mm from the disc and provided that the base is 6 - 15 mm diameter, ≤ 10 mm thick. Brachytherapy dose is 100 - 120 Gy delivered to the base and 40 Gy to the apex.
Protons may be used to provide better sparing of orbital structures and bones and improved lens sparing.
Toxicity of Treatment
Risk of second malignancy in familial Retinoblastoma with radiation is
- 4% at 10 years
- 18% at 35 years
- 50% at 50 years
Risk of second malignancy in familial retinoblastoma without radiation therapy is:
- 15 - 35% at 50 years.
- The second malignancies are mainly sarcomas, (soft tissue and osteosarcomas) and melanomas.
The risk of second malignancy in sporadic (non-familial) retinoblastoma is also increased but not as much as in familial retinoblastoma. The rate of second malignancies is 5% at 50 years.
Late effects of radiation include mid-facial hypoplasia, cataracts and second malignancies. Late effects of plaque brachytherapy are retinopathy, maculopathy, glaucoma and papillopathy.
Screening for trilateral retinoblastoma with bilateral retinoblastomas should be by MRI for at least 5 years biennually.