PEDS: Neuroblastoma
Neuroblastoma is a malignancy derived from the embryonic neural crest cells of the peripheral neural sympathetic nervous system. It is the most common extra-cranial solid tumor of childhood and accounts for 15% of all pediatric cancer related deaths. Neuroblastoma is marked by heterogeneous behavior, ranging from spontaneous maturation to rapid metastatic progression. Often advanced neuroblastom is the most lethal of childhood solid tumors, resisting all treatments.
Epidemiology, Screening and Genetics
There are about 650 cases in the US each year. The overall incidence is 10.5 /million population. The median age at diagnosis is 17 months.
The general age range of neuroblastoma is between birth to about 15 years. Less than 10% of neuroblastomas are found in those over 10 years. Adult neuroblastomas are quite rare and tend to have a slower growth rate than in pediatric populations.
Neuroblastoma accounts for around 10% of pediatritc cancer in children younger than 15 years making it the fourth most common malignancy of childhood, after leukemias, brain tumors and lymphomas.
Neuroblastoma is the most common malignancy of children under 18 months with an incidence of 29.1/million per year in children under 5 years. 90% are diagnosed in children younger than 10 years, 79% before 4 years and 36% are infants less than 1 year old. The median age at diagnosis is generally reported at 22 months (Halperin and Keole).
Anatomy
Neuroblastomas occur along the same distributions of the neural crest cells of the embryo. Embryonal neural crest cells, embryologically are found in columns dorso-lateral to the developing neural tissue.These columns are the embryologic precursors to the spinal root ganglia, the dorsal spinal nerves, and chromaffin cells which flank the abdominal aorta. The largest of these masses is the adrenal medulla. Most cases of neuroblastoma occur in a distribution consistent with these embryological origins.
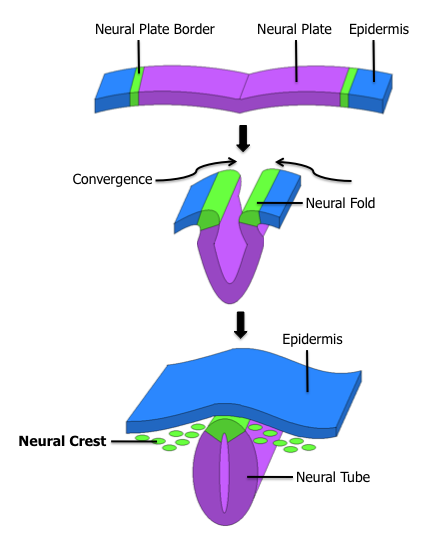
The general distribution of neuroblastoma are found:
- Cervical sympathetic ganglion: 1% (all), 4% (≤ 1 year old) 0.5% (age > 1 year old
- Posterior mediastinum: 19% (all) 29% (≤ 1 year) 14% (> 1 year)
- Adrenal Gland: 35% (all) 25% (≤ 1 year) 40% (> 1 year)
- Paraspinal ganglia in low thoracic, abdominal chains: 30%, 26% and 32%
- Pelvic paraspinal ganglia chains: 2%, 3% and 2.5 %
- Other sites: 12%, 31% and 9%
- Metastatic sites include bone, bone marrow, liver, lymphatics, skin. NEUROBLASTOMA RARELY METASTASIZES TO THE LUNGS. IF IT'S IN THE LUNGS, THEN IT'S NOT NEUROBLASTOMA! IT IS WILMS.
Neuroblastoma is a "small round blue cell tumor. Other small round blue cell tumors are Non-Hodgkin's Lymphoma, Ewing Sarcoma, undifferentiated soft tissue sarcomas including rhabdomyosarcoma, and PNET.
The classic histologic subtypes of neuroblastoma include:
- Neuroblastoma
- Ganglioneuroblastoma
- Ganglioneuroma
These tumors differ by the degree of cellular maturation.
There are three genetic syndromes in neuroblastoma. They are:
- Neurofibromatosis
- Hirchsprung Disease (congenital megacolon)
- Fetal hydantoin syndrome.
There are three neuroblastoma specific markers that distinguish neuroblastoma from other small round blue cell tumors:
- NSE
- synaptophysin
- neurofilament
N-myc amplification:
N-myc amplification is associated with a poor prognosis. N-myc amplification is identified by double staining chromatin bodies and homogenously staining regions. N-myc amplification is found in 30 - 40% of patients with neuroblastoma. There are three genetic/chromatin changes associated with poor prognosis neuroblastoma:
- N-myc amplification
- LOH 1p + 11q
- Diploid DNA
- Increased telomerase activity
DNA content does not have prognostic value in metastatic neuroblastoma.
70% of all neuroblastoma cases have 1p deletions.
Homozygosity for 3 single nucleotide polymorphisms on 6p22 are associated with Stage IV disease, n-myc amplification and disease relapse.
Screening
There have been multiple screening studies mostly centered around the testing for urine catecholamines (VMA, HVA). 90% of all neuroblastoma patients have detectable urinary catacholamines: vanillylmandelic acid or homovanillic acid. The most referenced study was a Quebec project which screened infants and compared the outcomes with case control cohorts of unscreened children in the remainder of Canada, Minnesota and Florida. The Quebec project increased the detection rate of neuroblastomas, but failed to have an impact on mortality in the screened populations. The reasons for this are a characteristic unique to neuroblastoma. It has a demonstrated ability to spontaneously regress. Screening practices detected almost exclusively biologically favorable disease (N-MYC non amplified, triploid, favorable Shimada histology). The conclusion is that screening practices resulted in overdiagnosis of tumors that would spontaneously regress. The screening studies area also hampered by the high rate of false positives.
Diagnosis: Signs and Symptoms
Along with the presentation of a mass, neuroblastoma may have other presenting signs. Neuroblastoma patients are sick patients. They have associated constitutional symptoms such as fever, malaise, pain and weight loss, periorbital ecchymosis (raccoon eyes, blueberry muffin sign which are blue non-tender skin metastases. Also presenting signs include bone pain, scalp nodules, irritible/ill appearance, diarrhea associated with increased production of paraneoplastic vaso-intestinal peptide production, Horner syndrome, opsomyoclonus truncal ataxia (rare. ataxis, random eye movements, myoclonal jerking associated with early stage and persisting after cure). Diarrhea and low potassium (Kerner-Morrison Syndrome) are also found with neuroblastoma.
The most common sites of disease are found where the most common sites of neural crest cells in the embryo were found:
- adrenal medulla
- paraspinal regions
- posterior mediastinum
Neuroblastoma rarely metastasizes to the lung. If there are lung mets, the disease is far more likely to be Wilm's disease. Neuroblastoma commonly metastasizes to the bones (in 50% of the cases.) followed by lymph nodes (in 35%), then bone marrow, liver, skin and orbits. Repeating: lung metastases are rare.
Bone metastases are common. The most common sites of bony metastases are the bones of the skull and orbit. Orbital metastases are the proximate cause of the raccoon eyes and proptosis.
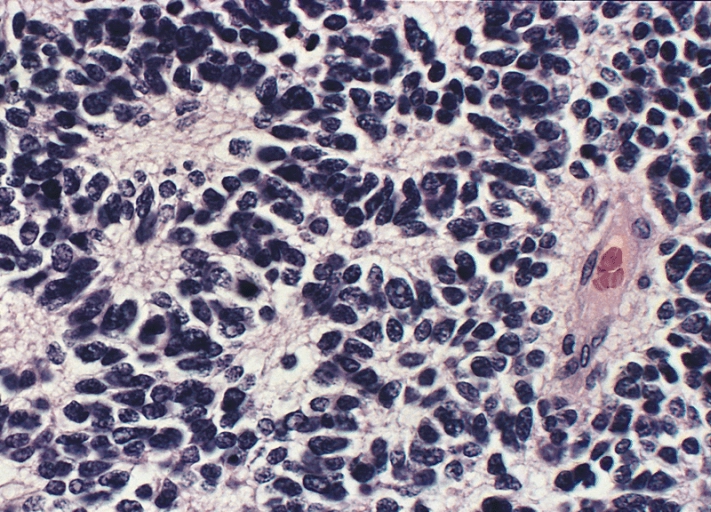
Neuroblastomas are more likely to exhibit calcifications. The classic histopathologic findings are Homer-Wright pseudorosettes, hemorrhage, and calcification.
| Adrenal Homer-Wright pseudorosette |
|---|
 |
Workup and Staging
Staging systems currently used in neuroblastoma in use by most cooperative groups include the International Neuroblastoma Staging System which describes the extent of resection. A newer staging system using only pre-treatment information has been developed and is being used in tandem with the INSS, the International Neuroblastoma Risk Group. The concurrent use of these two systems allows inter-trial comparisons.
International Neuroblastoma Staging System
| Stage 1 | unilateral localized tumor with GTR ± microscopic residual disease; ipsilateral lymph nodes negative |
| Stage 2A | Localized unilateral tumor with subtotal resection; no lymph nodes |
| Stage 2B | Localized tumor (GTR/STR) Ipsilateral lymph nodes positive, contralateral lymph nodes negative |
| Stage 3 |
|
| Stage 4 | Metastatic to distant lymph nodes, bone, bone marrow, liver. For children < 12 months see 4S |
| Stage 4S | Localized primary tumor (Stage 1-2B) age < 1 year; dissemination limited to skin, liver or bone marrow (less than 10% BM involvement) |
The International Neuroblastoma Risk Group
The International Neuroblastoma Risk group criteria divides local regional tumors and metastatic tumors. The local-regional tumors are scored L1 or L2 based on the absence or presence of 1 or more of 20 radiologic findings. These image defined radiologic findings generally describe whether or not a neuroblastoma tumor might be resectable. The authors of the INRG avoided terms like "resectable" to control for surgeon's judgement and style.
For metastatic disease, the INRG stage is defined as M, except for MS which are metastatic tumors confined to the skin, liver,and bone marrow (involving less than 10% total bone marrow) in patients < 18 months old.
Bones are not included in the INRG staging or the INSS staging "4S/MS" category. Only the bone marrow is included. the "4S/MS" category for bone marrow, liver and skin only includes those under 12 for INSS and 18 months for INRG. For patients older than 12/18 months, the proper stage is 4 or M.
Prognostic Factors in Neuroblastoma
The INRG looked at a database of 8800 neuroblastoma patients for the presence of prognostic factors. They found that patients could be stratified by risk/prognostic factors in to four groups based on 5 year event free survival. These prognostic factors include:
- Age
- Stage
- histologic category
- grade
- n-myc amplification
- 11q aberration
- DNA ploidy
The two clinical factors most predictive of cure are age (< 1 year gives the best prognosis, even over n-myc amplification) and Stage at Diagnosis.
Stage strongly influences prognosis with 90% Stage 1 and 2 survivors, 85% stage 4S survivors (skin, bone marrow, liver age ≤ 12 months). Stage 3 survival drops to 40% - 90% with multimodality therapy and Stage 4 is a dismal 40%.
Age < 12 months has been the traditional cutoff for good survival in the INSS but the INRG has shown that for favorable histology, age &1e; 18 months is likely to be favorable. Infants and toddlers without n-myc amplification have more than 80% long term survival with chemotherapy. Children age > 18 months and Stage 4 have only 40% long term survival.
COG Risk Groups
COG stratified neuroblastoma patients into low, intermediate and high risk groups using five factors:
- Stage, INSS (Stage 3 and 4 worse)
- Age (older is worse)
- N-myc status (amplified is worse)
- DNA ploidy
- Shimada Classification
The Shimada classification scheme divides neuroblastoma into favorable histology and unfavorable histology. The favorable factors are:
- Stroma-rich (Schwann cell stroma)
- Age (young)
- Differentiation (well differentiated is better)
- Mototic/karyorrhectic index (low)
- Nodularity (non-nodular is better)
TheCOG risk groupings are complex, General information follows:
- Low Risk:
- All INSS Stage 1 disease at any age is considered low risk.
- All INSS Stage 1, 2, and 4S disease and age < 1 year are low risk
- INSS Stage 2 and age > 1 year is low risk if favorable histology ± n-myc amplification or any histology and n-myc not amplified.
- Stage 4S and age < 1 year and favorable histology, n-myc not amplified and DNA index > 1
- Intermdiate Risk:
- Stage 3, age < 1
- Stage 3 age 1 - 21 n-myc normal, favorable histology
- Stage 4, age < 1, n-myc normal
- Stage 4S, age < 1, n-myc normal, and have either unfavorable histology or diploid DNA
- High Risk:
- Stage 2, age 1 - 21, n-myc amplified, unfavorable histology
- Stage 3, age < 1, n-myc amplified
- Stage 3, age 1 - 21 years, either n-myc amplified or unfavorable histology
- Stage 4 age < 1 and n-myc amplifies
- Stage 4S age < 1 and n-myc amplified
All Stage I neuroblastomas are considered low risk. There are no intermediate risk patients with n-myc amplification. These patients are either Stage 1, or Stage 2, 4S and age < 12 months low risk or they are high risk. The COG neuroblastoma patients are mostly high risk patients at 55%. Low risk patients account for only 30% of the diagnoses.
Stage 4S patients are intermediate risk if they do not have n-myc amplification and have either unfavorable histology or diploid DNA.
N-myc amplified patients are either low risk (Stage 1) or high risk (all other stages).
The OS-3 for neuroblastoma is segmented by risk group.
- Low Risk: 95% - 100%
- Intermediate Risk: 75 - 98%
- High Risk: < 30%
Staging Workup
The staging work up in suspected neuroblastoma cases includes the usual H&P, laboratory studies including urinary catecholamines, imaging studies as follows:
- bone scan
- CT chest abdomen and pelvis
- MRI liver abdomen and spine
- I-131 MIBG scan
- Bone marrow biopsy
- Pathologic studies for n-myc amplification, DNA content and cytogenetics.
Bone marrow biopsy is important in the workup of neuroblastoma as it helps establish stage. Bone marrow disease may eliminate the need for surgery at the primary site.
Treatment
Treatment regimens are segregated by risk groups.
Low Risk Groups
For low risk groups the present COG protocol is to treat with surgery alone with chmotherapy reserved for persistent or recurrent disease.
For low risk Stage 4S disease, biopsy and then supportive care. Chemotherapy and radiation are reserved for rapidly growing or symptomatic disease. This protocol was identified from subgroup analysis of the CCG 3881 study which showed that supportive care is sufficient for 57% of the patients. The EFS-5 was 86% and OS-5 92%.
The use of observation with out resection in infants with localized neuroblastoma is supported by German GPOH trials NB95-S and NB-97 for patients without n-myc amplification. In 93 patients with neuroblastoma, 44 had spontaneous regression. Overall survival and distant metastases free survivals were no different than those treated aggressively with surgery or chemotherapy. OS-3 99% and DM-FS-3 94%.
Intermediate Risk Groups
For intermediate risk groups, the treatment is surgery → short or long course chemotherapy depending on biolog factors ± 2nd look surgery if needed.
Radiation therap in intermediate risk groups is usually reserved to those who are not responding to initial chemotherapy and have bulky disease, such as those with respiratory distress due to hepatosplenomegally or spinal cord compression causing neurologic compromise. Radiation therapy is not indicated as consolidation therapy even in patients with persistent disease.
Indications for Radiation based on A3961:
- palliation
- viable residual disease in treatment refractory patients
- Recurrent disease
Radiation to the local area is 24 Gy.
For unfavorable Stage 4S disease (intermediate risk) the standared treatment is 8 cycles of chemotherapy.
High Risk Groiups
The general treatment paradigm is induction chemotherapy then surgical resection, then high dose chemotherapy followed by stem cell transplant followed by consolidation radiation therapy followed by oral cis-retinoic acid.
The COG 9532 protocol treats high risk patients with radiation to the post chemotherapy, pre-operative tumor bed to a total dose of 21.6 Gy at 1.8 Gy/fraction. If there is gross residual disease, this is followed by a 14.4 Gy boost to a total dose of 36.0 Gy.
Radiation therapy is not given electively to nodes in high risk disease. Nodes are included only if clinically or pathologically positive.
A secondary analysis of COG 3891 high risk neuroblastoma study (Haas-Kogan 2003 IJROBP) found that high risk neuroblastoma patients who received 10 Gy local EBRT and 10 Gy TBI as part of a transplant program had better local control than patients who did not get TBI or a transplant. The LRR-5 was 22% (in favor of TBI) compared with 52% for those who did not. These results are used to support the current 21.6 Gy dose in high risk protocols.
CCG 3891(Matthay 2009 JCO) treated high risk neuroblastoma patients with induction chemotherapy → surgery and 10 Gy to gross residual disease. Patients were then randomized to 3 cycles of non-myeloablative chemotherapy, TBI and bone marrow transplant. Patients underwent secondary randomization to observation v. cis-retinoic acid x 6 months. Both chemotherapy and cis-retinoic acid improved OS-5. OS-5 for patients who received both was 59%.
Chemotherapy
Chemotherapy drugs typically used in neuroblastoma are:
- cytoxan
- doxorubicin
- etoposide
- carboplatin
- ifosfamide
Radio-isotope Therapy
131I MIBG can be used for refractory neuroblastoma based on a promising Phase II study showing a 36% response rate. (Matthay et al 2007 JCO).
Emergencies: Cord Compression and Hepatosplenomegaly
For symptomatic spinal cord compression treat:
- If age < 3 years old treat to 9 Gy at 1.8 Gy/fraction
- If age ≥ 3 years old, treat to 21.6 Gy at 1.8 Gy/fraction
For symptomatic hepatosplenomegaly treat to 4.5 Gy at 1.5 Gy x 3 fractions.
Toxicities
Dose Constraints
Contralateral Kidney: Limit dose to the entire contralateral kidney to < 15 Gy
Liver V15 is to be held to < 66%
Lung V15 is to be held to < 66%